Résumés CPLF 2026
Approche hybride pour le dépistage des formes atténuées du déficit en sphingomyélinase acide de type B
Auteur correspondant : Villeneuve T.
Introduction
Le déficit en sphingomyélinase acide (ASMD) de type B est une affection lysosomale rare, secondaire à des mutations du gène SMPD1 . Sa faible prévalence et l’hétérogénéité de ses phénotypes cliniques compliquent le diagnostic, alors que la disponibilité récente d’une thérapie enzymatique substitutive confère un intérêt majeur au dépistage précoce [1,2].
Méthodes
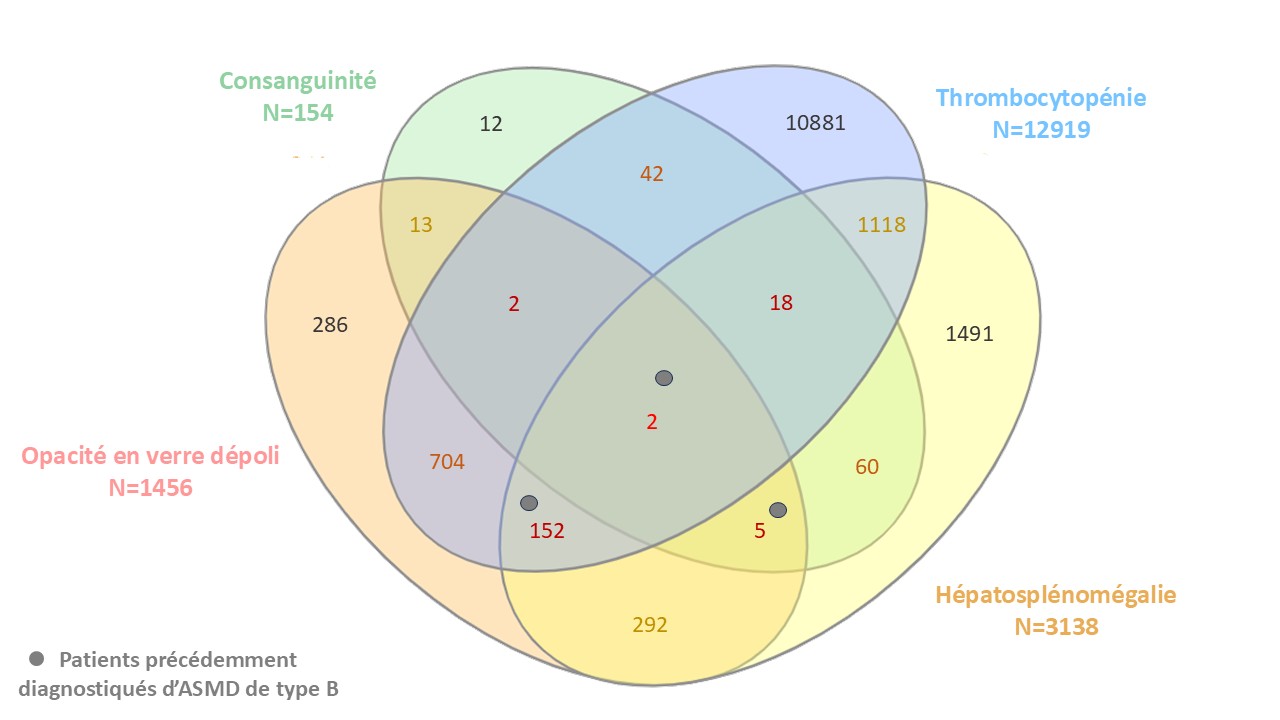
Nous avons conduit une étude rétrospective monocentrique (RnIPH 2024-85) au CHU de Toulouse, portant sur 359 802 bilans lipidiques réalisés entre 2012 et 2023. Les patients présentant un rapport cholestérol total/HDL>4,5 ont été sélectionnés. Un algorithme d’extraction automatisée de données cliniques a été appliqué afin de repérer la consanguinité, l’hépatomégalie, la splénomégalie et la présence d’opacités en verre dépoli. La thrombopénie (< 150 × 10⁹/L) a également été intégrée comme critère. Les patients remplissant au moins quatre critères sur cinq ont ensuite bénéficié d’une relecture clinique approfondie.
Résultats
Parmi 63 653 patients avec un rapport cholestérol total/HDL>4,5, 20,3% présentaient une thrombopénie, 4,93% une hépatosplénomégalie, 2,29% des opacités en verre dépoli et 0,24% un antécédent de consanguinité. Cent soixante-dix-neuf patients avaient≥4/5 critères, dont 19 (10,6%) étaient pédiatriques. Les 3 cas déjà connus d’ASMD de type B ont été identifiés. Par ailleurs, 46 patients (25,7%) présentaient une maladie monogénique identifiée et 5 patients sans diagnostic sont relevables d'un dépistage spécifique de l’ASMD de type B.
Conclusion
Cette stratégie de dépistage, associant extraction automatisée de données et validation clinique, a permis d’identifier efficacement des cas avérés et suspects d’ASMD de type B. L’approche hybride proposée illustre l’apport des outils informatiques dans l’amélioration du diagnostic des maladies rares et pourrait contribuer à un accès plus précoce au test génétique.
Références
[1] Lachmann, R. H. et al. Olipudase alfa enzyme replacement therapy for acid sphingomyelinase deficiency (ASMD): sustained improvements in clinical outcomes after 6,5 years of treatment in adults. Orphanet J Rare Dis 18, 94 (2023).
[2] Diaz, G. A. et al. One-year results of a clinical trial of olipudase alfa enzyme replacement therapy in pediatric patients with acid sphingomyelinase deficiency. Genetics in Medicine 23, 1543–1550 (2021).
Villeneuve T. Sanofi Genzyme ; Jamme T. Sanofi Genzyme ; Schwob R. * ; Levade T. Sanofi Genzyme ; Prévot G. Sanofi Genzyme
*Déclare ne pas avoir de lien d'intérêt en rapport avec ce résumé.
